
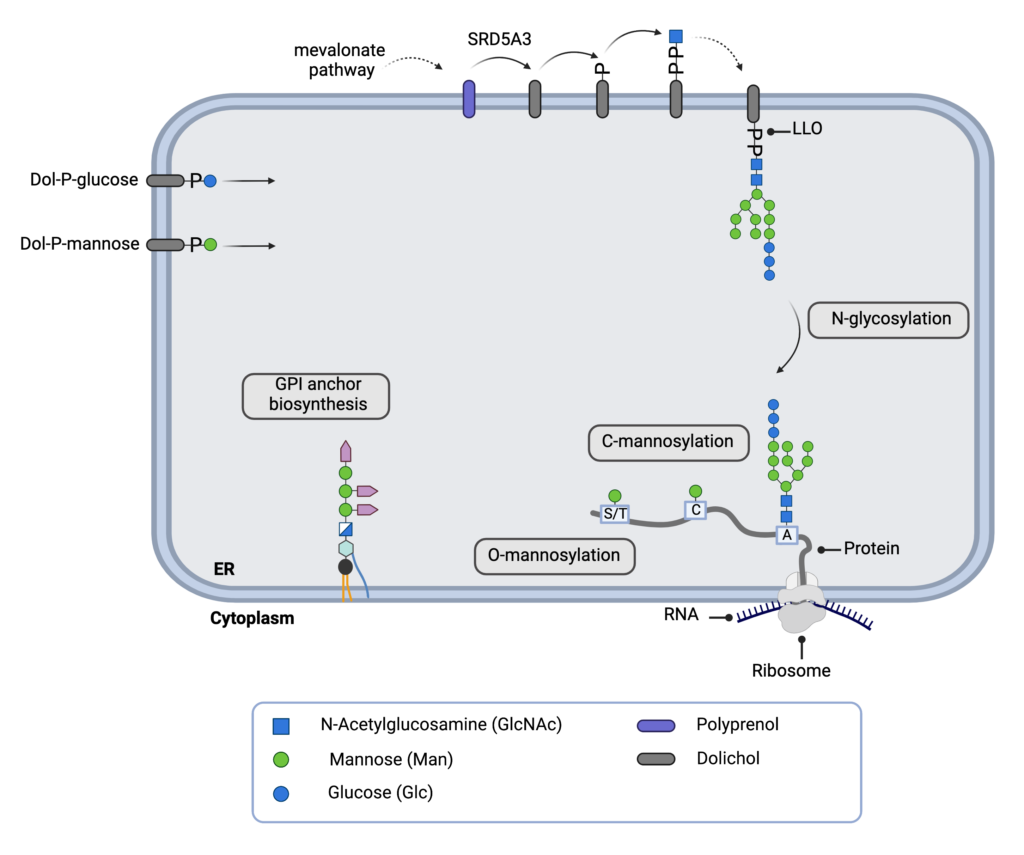
RD5A3-CDG, formerly known as CDG-Iq, is a rare inherited condition that affects several body systems. To date, approximately 50 cases of SRD5A3-CDG have been reported in the literature. SRD5A3-CDG is classified as a disorder of multiple glycosylation pathways and can be subcategorized as a disorder of dolichol metabolism. It is caused when an individual has mutations in both copies of the SRD5A3 gene, which provides instructions for making an enzyme that converts the molecule, polyprenol, into dolichol – a lipid that is needed for different types of glycosylation. Mutations in the SRD5A3 gene reduce the availability of dolichol, leading to proteins and lipids with abnormal sugar chains. Symptoms of SRD5A3-CDG begin in infancy and commonly include neurological abnormalities (problems with balance and coordination, low muscle tone and intellectual disability) vision loss early on in life, eye abnormalities, unusual facial features and skin symptoms. Some individuals may present with heart and liver problems and feeding difficulties. A screening test is available for SRD5A3-CDG but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for SRD5A3-CDG but gene therapy and drug repurposing are currently being explored in pre-clinical studies. Current treatment focuses on managing specific symptoms and preventing complications.
For more details, click here
SRD5A3-CDG is a rare, non X-linked congenital disorder of glycosylation due to steroid 5 alpha reductase type 3 deficiency characterized by a highly variable phenotype typically presenting with severe visual impairment, variable ocular anomalies (such as optic nerve hypoplasia/atrophy, iris and optic nerve coloboma, congenital cataract, glaucoma), intellectual disability, cerebellar abnormalities, nystagmus, hypotonia, ataxia, and/or ichthyosiform skin lesions. Other reported manifestations include retinitis pigmentosa, kyphosis, congenital heart defects, hypertrichosis and abnormal coagulation.
For more details click CLICK HERE
The protein encoded by this gene belongs to the steroid 5-alpha reductase family, and polyprenol reductase subfamily. It is involved in the production of androgen 5-alpha-dihydrotestosterone (DHT) from testosterone, and maintenance of the androgen-androgen receptor activation pathway. This protein is also necessary for the conversion of polyprenol into dolichol, which is required for the synthesis of dolichol-linked monosaccharides and the oligosaccharide precursor used for N-linked glycosylation of proteins. Mutations in this gene are associated with congenital disorder of glycosylation type Iq.
For more details, click here
Synonyms: Congenital Disorder of Glycosylation Type Iq, Coloboma, Ocular, with Ichthyosis, Brain Malformations, and Endocrine Abnormalities, CDGIq, Congenital disorder of glycosylation due to steroid 5alpha-reductase type 3 deficiency, CDG-Iq, Congenital disorder of glycosylation type 1q, Congenital disorder of glycosylation type Iq, SRD5A3-CDG, CDG syndrome type Iq.
For more details, click here.
SRD5A3 plays a key role in early steps of protein N-linked glycosylation by being required for the conversion of polyprenol into dolichol. Dolichols are required for the synthesis of dolichol-linked monosaccharides and the oligosaccharide precursor used for N-glycosylation. Acts as a polyprenol reductase that promotes the reduction of the alpha-isoprene unit of polyprenols into dolichols in a NADP-dependent mechanism. Also able to convert testosterone (T) into 5-alpha-dihydrotestosterone (DHT).
For more details click here
SRD5A3-CDG (also known as CDG syndrome type Iq, CDG-Iq, CDG1Q or Congenital disorder of glycosylation type 1q) is a rare, non X-linked congenital disorder of glycosylation (CDG)due to a mutation in the steroid 5 alpha reductase type 3 gene. It is one of over 150 documented types of Congenital disorders of Glycosylation. Like many other CDGs, SRD5A3 is ultra-rare, with around 38 documented cases in the world.
For more details click here